initialize and repopulate git
parents
BulkRNAseq/.gitkeep
0 → 100644
BulkRNAseq/00-Interaction.R
0 → 100644
File added
{kind=link}
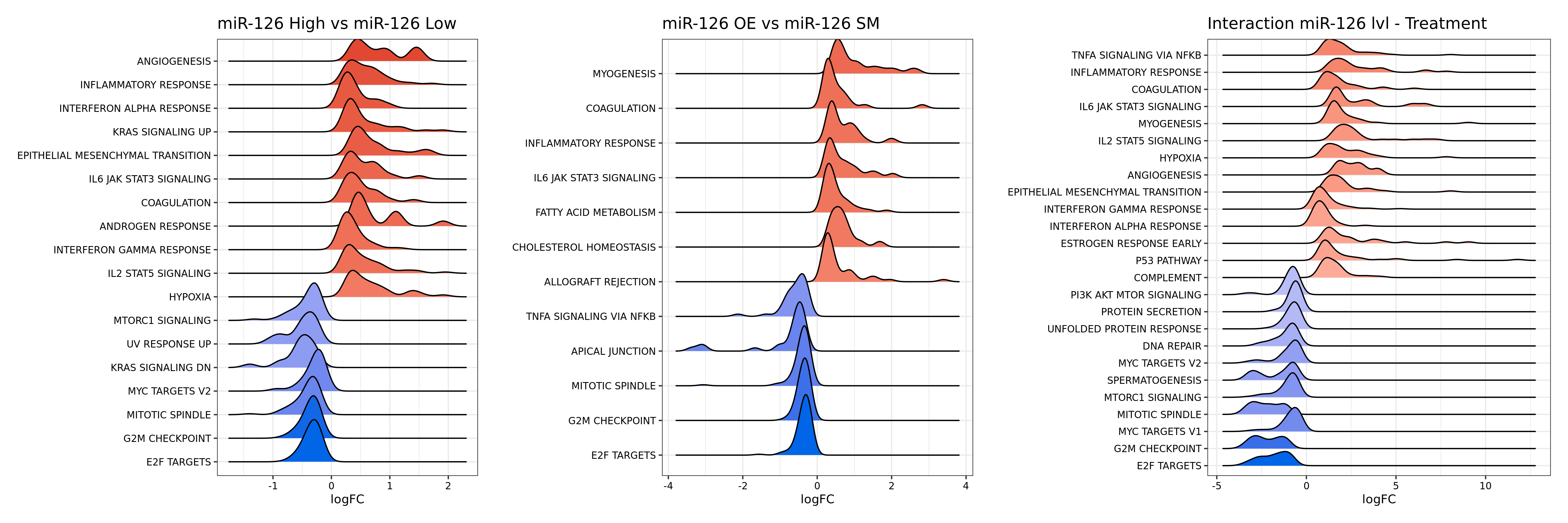
675 KB
BulkRNAseq/README.md
0 → 100644
Methylation/.gitkeep
0 → 100644
Methylation/README.md
0 → 100644
README.md
0 → 100644
WES/.gitkeep
0 → 100644
scRNAseq/.gitkeep
0 → 100644
{kind=link}
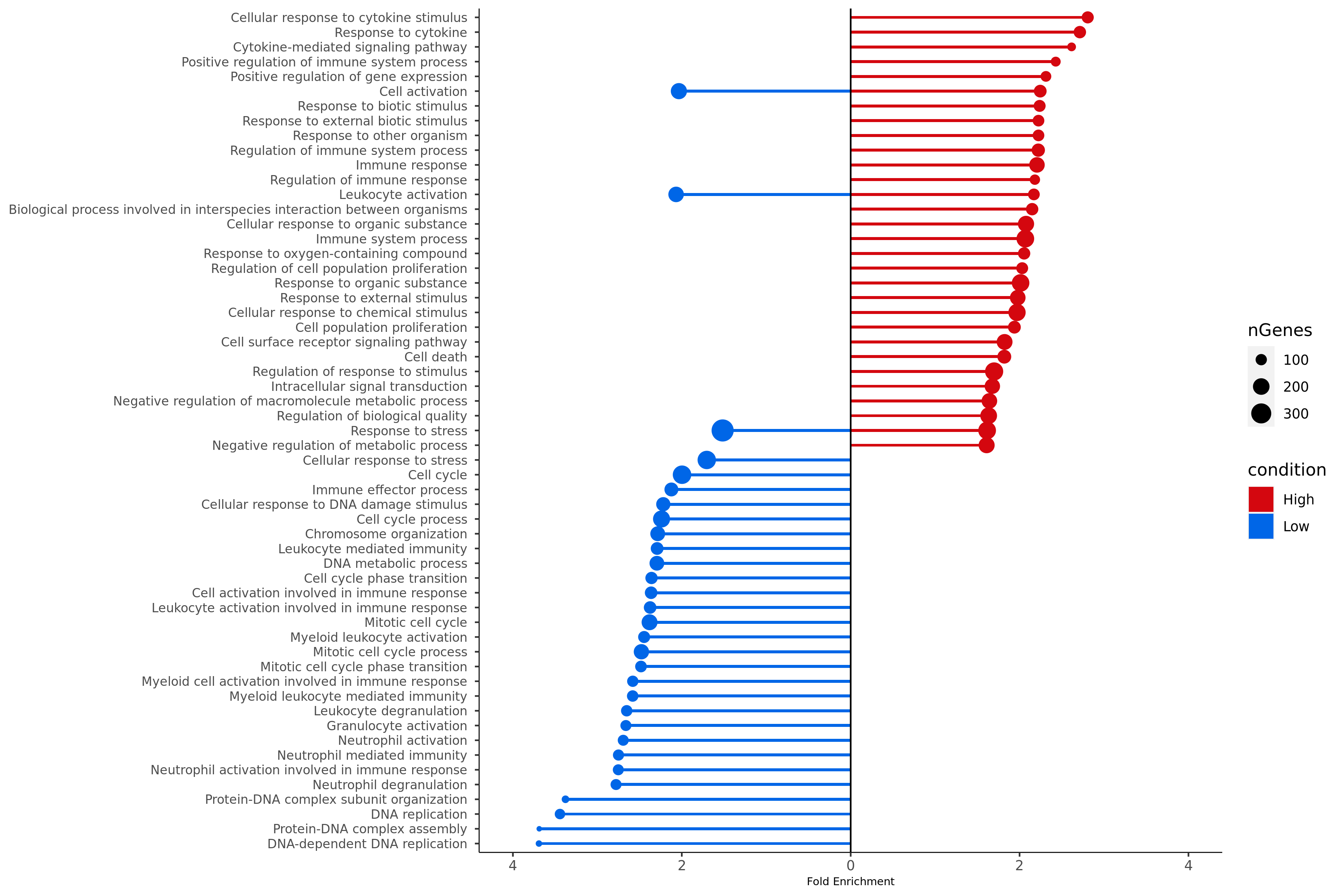
440 KB
{kind=link}
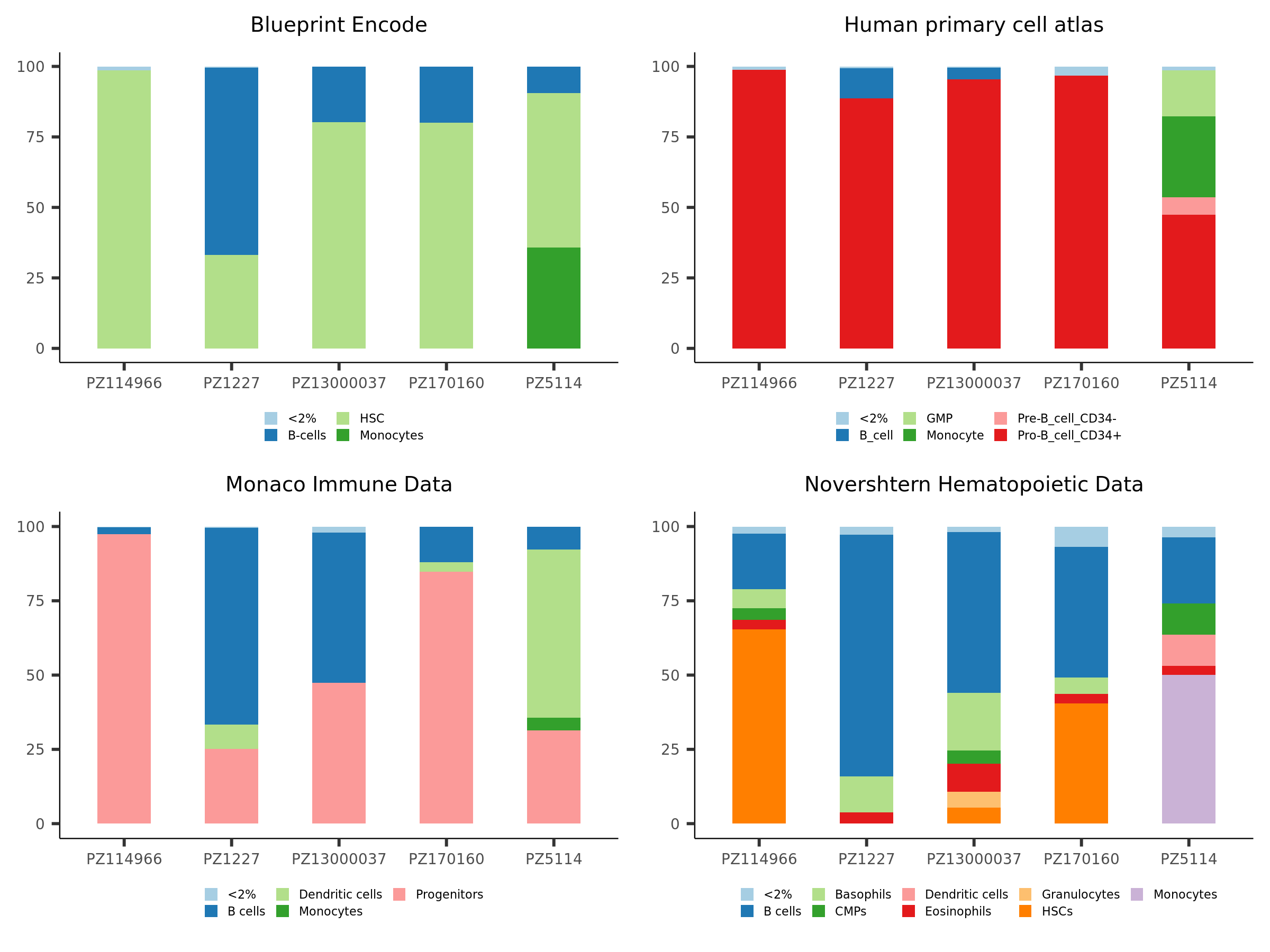
83.1 KB
scRNAseq/PaperPanels.R
0 → 100644
This diff is collapsed.
scRNAseq/README.md
0 → 100644
scRNAseq/high_GObiopro.csv
0 → 100644
This diff is collapsed.
scRNAseq/low_GObiopro.csv
0 → 100644
This diff is collapsed.